
Anu Suomalainen Wartiovaara
Forskningsprogrammet för molekylär neurologi
Haartmansgatan 8
00290 Helsingfors
anu.wartiovaara [at] helsinki.fi
09-471 71965
Patient information
1. Vad är mitokondrier?
2. Vad är mitokondriesjukdomarna?
3. Vad är mitokondrieDNA?
4. Vad händer om det finns genfel i mitoDNA?
5. Hur ärvs mitoDNA?
6. Ärvs alla mitokondriesjukdomar från modern?
7. Vad är multipla deletioner av mitoDNA?
8. Hur undersöks mitkondriernas funktion vid misstanke om en mitokondriesjukdom?
9. Blir man alltid sjuk om man har genfel i mitoDNA?
10. Om jag har en släkting som diagnostiseras med en mitokondriesjukdom, var får jag information om hur fyndet påverkar mitt liv?
11. Kan mitokondriesjukdomspatienter med höga kolesterol- och fettvärden använda statinmedicinering för att sänka kolesterolhalten?
Mitokondrier är organeller inne i cellen. De är cellens små energikraftverk. Deras uppgift är att förvandla födans energi till en sådan form som kroppens vävnader kan utnyttja. Den här processen använder också en stor del av syret vi inandas. Således hamnar både näringsämnen från maten och syret i andningsluften i mitokondrierna, och blir råvaror för deras energiproduktion.
Mitokondrierna har även andra funktioner utöver energiproduktionen. De deltar i syntesen av hemgrupper och vissa hormoner, samt i lagringen av kalcium.
2. Vad är mitokondriesjukdomarna?
Mitokondriesjukdom är ett samlingsnamn för ett antal olika sjukdomar vars gemensamma nämnare är rubbad mitokondriefunktion. Symptomen antas bero på otillräcklig energimetabolism. De svåraste formerna av mitokondriesjukdomar kan visa symptom snart efter födseln och leda till spädbarnets död. Andra mitokondriesjukdomar ger symptom först vid hög ålder och är inte nödvändigtvis livshotande eller inverkande på patientens livslängd. Mitokondriesjukdomarnas variation är så stor, att man kan finna en för varje åldersgrupp och nästan varje medicinsk specialitet. Energibristen drabbar oftast muskler, hjärta och/eller hjärna, och hos barn även levern. Också vissa hörselfel är typiska symptom för mitokondriesjukdomar, speciellt om de förekommer i samband med diabetes, antingen hos samma patient eller om det finns mycket diabetes i dennes släkt.
3. Vad är mitokondrieDNA?
Generna styr alla cellfunktioner, så även mitokondriernas funktion. Största delen av mitokondriernas proteiner produceras av gener som finns i cellkärnan. Av dessa gener ärver vi hälften av fadern och hälften av modern. Mitkondrierna har även egen arvsmassa som kallas mitokondrieDNA, eller mito-DNA, och som behövs i vävnadernas och organens energiproduktion.
4. Vad händer om det finns genfel i mitoDNA?
Genfel i mitoDNA påverkar vävnadernas förmåga att producera energi. Vi känner till många olika typer av genfel i mitoDNA: stora deletioner (partiella brister) och genfel som berör bara enstaka baspar. Till exempel MELAS är en sjukdom som orsakas av ett genfel i mitoDNA. Felet är mycket litet: ungefär i samma storleksklass som en felaktig bokstav på 5 stycken A4-sidor text. Trots det är felet stort nog för att störa energiproduktionen.
5. Hur ärvs mitoDNA?
Det finns upp till hundratusen kopior av mitoDNA i en äggcell. Barnen får all sin mitoDNA från modern, fast allt annat genetiskt material kommer till hälften från fadern. Hur ett mitoDNA-fel ärvs beror på felets typ, som beskrivs nedan:
Punktmutationer (till exempel mutationer relaterade till sjukdomrarna MELAS, MERRF, NARP). Om en del av mitoDNA:t i en äggcell innehåller genfel, ärver avkomman dessa felaktiga kopior av mitoDNA. Om den befruktade äggcellen innehåller t.ex. mitoDNA med MELAS-genfel, ärver avkomman felet. Ifall äggcellens alla kopior av mitoDNA bär MELAS-genfelet kan den inte befruktas eftersom energibristen är för stor. Därför har alla barn till mammor med MELAS-genfel både normalt mitoDNA och mitoDNA med MELAS-felet.
Eftersom mitoDNA ärvs vidare enbart från modern, har sjuka män ingen risk att ge vidare sina mitoDNA-fel till sina barn. Det är inte heller givet att barn till sjuka mammor faktiskt blir sjuka: ifall barnet har ärvt bara en liten mängd mitoDNA med MELAS-fel, kan det hända att sjukdomen aldrig bryter ut.
stora enskilda deletioner av mitoDNA betyder att en del av patientens mtDNA-molekyler saknar stora bitar. Dessa förekommer t.ex. i mitokondriella muskelsjukdomar, en del av de mitokondriella sjukdomar som orsakar svaghet i ögonmusklerna, samt i Pearsons syndrom och Kearns-Sayres syndrom. Stora enskilda deletioner av mitoDNA är vanligtvis inte ärftliga. Det är sannolikt att sådana deletioner har uppstått under patientens tidiga fosterutvecklig istället för att vara nedärvda av modern. Därför är det osannolikt att en mor vars ena barn har en stor enskild deletion skulle löpa risken att få ett barn till med samma problem. Det är också sannolikt att en kvinnlig patient med en enskild stor deletion inte kommer att ge felet vidare till sina eventuella barn.
6. Ärvs alla mitokondriesjukdomar från modern?
Svaret är nej. Av mitokondriesjukdomar är det bara punktmutationer som ärvs enbart från modern åt alla barn. Mitokondriernas funktion påverkas av ett stort antal gener i cellkärnan, och dessa ärvs ju till hälften av fadern. I dessa fall kan nedärvningsmöstret vara antingen dominant (avkomman har 50% risk att ärva en felaktig gen av sin sjuka förälder) eller recessivt (avkomman har 25% risk att få sjukdomen, eftersom det krävs en felaktig kopia av båda föräldrarna).
7. Vad är multipla deletioner av mitoDNA?
Med åldern kan det ansamlas fel i mitoDNA. Det beror på att patienten har fel i ett protein som sköter uppehället av mitoDNA. Dessa proteiner byggs alla enligt anvisningar från generna i cellkärnan, och har således ärvts av båda föräldrarna. Del i de gener som behövs för att producera proteinerna Polymeras gamma (POLG) och Twinkle är de vanligaste orsakerna till den här typen av ansamling av fel i mitoDNA. POLGs och Twinkles uppgift är att kopiera och reparera mitoDNA. Till exempel då cellen delar på sej måste också arvsmassan dupliceras. Om det finns sådana fel i POLG eller Twinkle att de inte klarar av att ordentligt kopiera och duplicera mitoDNA, blir kopiorna felaktiga med tiden. Typiska följder av POLG och Twinkle –genfel är många partiella stora brister i mitoDNA, d.v.s. så kallade multipla deletioner. Sådana deletioner ligger bakom t.ex. ärftlig ögonmuskelsvaghetssjukdom (PEO), och sjukdomen MIRAS vars symptom är bl.a. degeneration av lillhjärnan och degenerationer i perifera nervsystemet. Om en provbit av patientens muskel visar multipla deletioner i mitoDNA eller nedsatt antal mitoDNA-molekyler, sökes genfelet i första hand i POLG-genen. Man skall dock komma ihåg att förekomsten av ett mindre antal fel i mitoDNA är en del av normalt åldrande, och utgör inte ensamma grund för diagnos av en mitokondriesjukdom.
8. Hur undersöks mitkondriernas funktion vid misstanke om en mitokondriesjukdom?
Första steget är naturligtvis att läkaren kommer att tänka på en mitokondriesjukdom som förklaring till patientens symptom. Om patienten har hjärn-relaterade symptom, kan magnetröntgen av huvudet vara nyttigt. Avvikande röntgenfynd i hjärnan är typiska för mitokondriesjukdomar.
Laboratorieprov: Man kan få en uppfattning om mitokondriernas verkningsgrad genom att mäta energimetabolismens användning av syre och ansamling av slut- och biprodukter. Eftersom mitokondrier använder syre som råvara, indikerar stor syreanvändning effektiv mitokondriefunktion. Riklig produktion av koldioxid indikerar samma sak, eftersom koldioxiden är energimetabolismens slutprodukt. Om det finns problem med cellernas energiproduktion kan det uppstå laktat (mjölksyra) som biprodukt. Laktatproduktionen beror på cellernas alternativa sätt att producera energi utan mitokondrier. Den här processen sker utan syre, och laktat är en biprodukt av processen. Om mitokondriernas energiproduktion är bristfällig ökas mängden laktat i vävnader och blodet. Laktatmängden kan mätas från blod eller rygmärgsvätska. Höjda laktatvärden indikerar en mitokondriesjukdom, men å andra sidan orsakar alla mitokondriesjukdomar inte ökade laktatvärden. Ibland kan en tillfällig höjning av laktatvärdena förekomma i samband med hjärnsymptom, vilket kan konstateras med magnetröntgen kombinerat med spektroskopi. Ökning av laktatvärden kan också testas med en konditionscykelstest, där laktatvärdena registreras både under och efter ansträngning. Samtidigt kan man göra en spiroergometriundersökning som registrerar användning av syre och produktionen av koldioxid. Då mitokondrierna inte fungerar för fullt, används mindre syre och produceras mindre koldioxid är normalt. Om patienten har tydliga muskelsymptom, kan det hända att kreatinkinas-värdena (CK) är höjda som följd av en muskelsjukdom. Vanligtvis stiger CK inte till tusenden i mitokondriesjukdomar, förutom i svåra fall av muskeldystrofi-typens mitokondriesjukdomar hos barn.
muskelibiopsi. Muskelbiopsin är en väsentlig undersökning ifall man misstänker en mitokondriesjukdom. Vanligtvis tar kirurgen en provbit från t.ex lårmuskeln (musculus vastus lateralis) eller axelmuskeln (muscus deltoideus). Provbiten tas under lokal anestesi. Beroende på vilka prover man vill göra med provbiten, varierar bitens storlek från en motsvarande halva lillfingernageln till 1x3cm. Muskler är mycket olika och deras energiproduktion varierar, därför kan inte vilken muskel som helst användas för det här testet.
Biopsibiten kan sedan användas för att göra a) ett funktionellt test av mitokondrierna b) undersökning av mitoDNA och c) mikroskopisk undersökning av vävnaden.
- En funktionell undersökning av mitokondrierna innebär att man isolerar mitokondrierna från biopsibiten. För att få ett pålitligt resultat måste tiden mellan biopsin och undersökningen vara så kort som möjligt; helst under 4 timmar. Man mäter mitokondriernas förbrukning av syre och/eller deras energiproduktion genom att bjuda dem olika näringsämnen som de förbränner och producerar energi. Utöver detta undersöks sk. andningskedjans aktivitet. Till exempel MELAS orsakar brister i andningskedjans enzymkomplex I, vilket ofta syns i just den här analysen.
- Undersökning av mitokondrieDNA. Man isolerar DNA från muskelbiopsin, och använder provet för att söka defekter i mitoDNA. Det är typiskt för vissa mitokondriesjukdomar att visa tydliga tecken av mitoDNA:s fel i muskeln (t.ex. den ärftliga PEO-sjukdomen, reducerat antal mitoDNA-molekyler), emedan vissa sjukdomar inte visar några tecken alls (PEO, stora deletioner av mitoDNA) eller syns mycket svagare i vita blodkropparnas DNA (t.ex. MELAS, punktmutationer). I sådana fall är muskelbiopsin den enda möjligheten att konstatera genfelet.
- En mikroskopisk undersökning av vävnaden. En del av muskelbiopsin fryses ner och en patolog gör histologiska färgningar åt den. Sedan analyseras mitokondriernas funktion och antal med mikroskop. Om det syns så kallade COX-negativa muskelfibrer i vävnaden (se bilden), kan man konstatera att Cytokrom-c-oxidasens aktivitet har sjukit. Cytokrom-c-oxidas är en del av mitokondriernas energiproduktionsprocess. Med andra ord berättar COX-negativa fibrer om rubbningar i mitokondriernas funktion. Om man undersöker vävnadsprovet under ett elektronmikroskop ser man ofta ett stort antal mitokondrier som har avvikande form och storlek (se bilden).
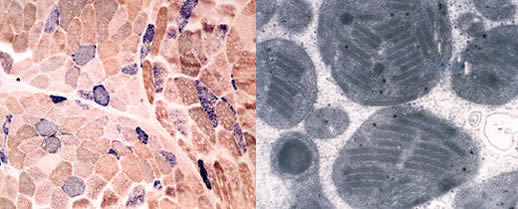
I bilden till vänster syns ett tvärsnitt av muskelfibrer. De blåfärgade fibrerna är COX-negativa, d.v.s. de lider av nedsatt aktivitet av andningskedjans COX-entsym och har ett ökat antal mitokondrier (bild A. Paetau). |
I bilden till höger en elektronmikroskopbild som visar förstorade mitokondrier som innehåller avvikande element, s.k. parkeringsplatselement.
|
9. Blir man alltid sjuk om man har genfel i mitoDNA?
När en patient har symptom som stämmer med en mitokondriesjukdoms sjukdomsbild och man hittar ett genfel i mitoDNA, är detta genfel orsaken till symptomen. Hos en frisk människa hittar man genfel i mitoDNA vanligtvis bara då man undersöker släktingar till en patient som har en mitokondriesjukdom och en från modern ärvd punktmutation i mitoDNA (t.ex. MELAS). När en patient diagnostiseras med ett sådant genfel gäller situationen plötsligt alla dennes släktingar på mödernet: syskon, modern, moderns syskon, systrarnas barn och en kvinnlig patients egna barn.
En enstaka cell har hundatals, även tusentals kopior av mitoDNA, så en enda felaktig kopia gör ingen skillnad för energiproduktionen. Det är antalet felaktiga kopior som är avgörande för utbrottet av sjukdomen. D.v.s. hur många bristfälliga kopior av mitoDNA fanns i den äggcellen man härstammar från. En patients biopsi kan till exempel visa att i de undersökta cellerna bär 70% av kopiorna MELAS-genfelet, medan 30% är felfria kopior.
Antalet felaktiga kopior avgör om vävnaden lider av energibrist eller inte. Om antalet felaktiga kopior av mitoDNA är litet, kan symptomens utbrott komma senare, manifestera sej mildare, eller helt låta bli att ge upphov till symptom. En människa kan således bära på MELAS-genfel utan att veta om det. I en och samma släkt kan man ha mitokondriesjukdomar av varierande svårhetsgrad, och man kan också hitta symptomfria släktingar från moderns sida. De bär nog på MELAS-mutationen, men den påverkar inte deras liv.
10 Om jag har en släkting som diagnostiseras med en mitokondriesjukdom, var får jag information om hur fyndet påverkar mitt liv?
Den centrala informationskällan är läkaren som sköter den sjuka släktingen. Hur fyndet påverkar andra i släkten beror helt på vilket genfel det är som ligger bakom sjukdomen. Om det gäller en punktmutation som ärvs av modern till alla barn (t.ex. MELAS), kan ett stort antal släktingar beröras av fyndet. Då kan den skötande läkaren skicka friska släktingar till en speliacist i medicinsk genetik för rådgivning.
Om det istället gäller en stor, enskild deletion av mitoDNA berörs släktingarna inte, eftersom det här felet inte är ärftligt.
Gäller det ett genfel i cellkärnans gener, kan specialisten i medicinsk genetik diskutera med släktingarna om hur fyndet påverkar deras liv, och hjälpa också med familjeplaneringen.
Tjänster av en specialist i medicinsk genetik fås åtminstone från universitetssjukhus, en del centralsjukhus och från Väestöliitto.
11 Kan mitokondriesjukdomspatienter med höga kolesterol- och fettvärden använda statinmedicinering för att sänka kolesterolhalten?
Vanligtvis rekommenderas mitokondriesjukdomspatienter att använda sådana kolesterolmediciner som påverkar enbart upptagningen av kolesterol, inte mitokondriernas funktion. Statinmedicineringens effekt baserar sej på att den hämmar entsymet HMG-coA reduktas, som påverkar produktionen av kolsterol. Samma entsym är dock en del av syntesen av mitokondriernas egna antioxidant ubikinon, som är en del av mitokondriernas andningskedja. Mitokondriesjukdomar försvagar ofta just andningskedjans funktion.
Eftersom statinerna hämmar ubikononets syntes rekommenderas mitokondriesjuksomspatienter att inte använda statiner.
Mitokondriesjukdomspatienternas vård planeras alltid individuellt, och då måste man väga fördelarna mot nackdelarna med medicineringen. Om man bestämmer sej för att använda statiner trots riskerna, är det viktigt att uppfölja patientens mående med regelbundna blodprov av speciellt CK-värden.
Fick du inte svar på din fråga? Om du tror att svaret på din fråga kunde vara av intresse och hjälp åt andra patienter och deras familj, kan du skicka din fråga till anu.wartiovaara [at] helsinki.fi